This curriculum will provide an informative, concise guide focused on the practical aspects of cryo-electron tomography or “cryo-ET” workflows. The technique provides a powerful tool to access morphological information and is the only method to provide structural and functional information of individual particles in their native environment. Cryo-ET encompasses multiple different workflows since its versatility allows for a broad range of samples, from large cells to single particles. The steps taken to successfully obtain results need to be adapted for each particular project and research question. CryoET 101 is divided into six chapters, each of which is focused on a different step in the cryo-ET pipeline.
If cryo-ET is the method to answer your research question, and you need training and/or access to specialized equipment, the National Network for CryoET (https://www.cryoemcenters.org/cryoet-centers/) is an excellent resource available to all.
Transmission electron tomography has traditionally been used to reveal cellular morphologies at an ultrastructural level using plastic-embedded stained samples and to study changes in cells and tissues during disease. More recently, however, breathtaking advances in cryo-electron microscopes, stages, direct electron detectors, energy filters, phase plates, and data processing software in combination with ongoing development of advanced sample preparation techniques of frozen hydrated cells and other specimens have been instrumental to allow for molecular-level views, some with near-atomic resolution.
This first chapter introduces the concept, basic principles and application of cryo-ET in three modules. Understanding the covered topics will help you make an informed decision if cryo-ET is a suitable method to answer your specific scientific question.
Tomography is a common technique that provides 3D information of an object and is used in many areas of medicine and science. For example, computed tomography (CT), positron emission tomography (PET) and magnetic resonance imaging (MRI) used in medicine are all comparable in concept to electron tomography: a two dimensional set of rotational images is collected from a 3D object. These 2D projections are subsequently transformed into a volume providing spatial information.

A difference lies in the scale: electron-tomography focuses on cellular and molecular level rather than whole organs or organisms. In fact, cryo-ET is the only method that can achieve near atomic resolutions of macromolecules and cellular structures directly observed in their native context. The resolution range for cryo-ET is large and it bridges micron level resolutions where ultrastructural features can be observed to nanometer resolutions, where macromolecules and domain level detail within proteins can be discerned. If multiple copies of a molecule can be identified, they can be averaged to obtain atomic resolution maps. More commonly, resolutions of 15-30 Å are achieved. Molecules of interest need not be isolated or purified as they do for other common structural methods like single particle cryo-EM and X-ray crystallography. Using in situ cryo-ET, the study of large molecular machines, their assemblies and interaction partners within their native compartments become accessible and can be observed in different conformational and functional states. In addition, biological processes such as changes in cells in response to stress or injury or virus replication and egress can be directly visualized.
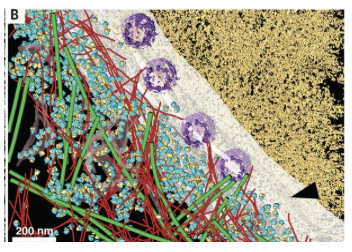
Cryo-ET is used to obtain 3D information of a frozen vitrified sample. The advantage of cryo-ET is that it can be applied to many different samples – from molecules, to complexes, viruses, bacteria, cells, tissues, small organisms and more. However, the caveat is that the sample needs to be vitrified and sufficiently thin to be penetrated by the electron beam, which may require additional sample preparation procedures. For samples that are too thick, a ‘thinning’ step is included in the sample preparation, such as slicing with an ultra-microtome or using a focused ion beam (FIB) to produce thin cryo-lamella. In addition, molecules or structures that are fluorescently tagged can be correlated to the final tomograms through a process called cryo-correlated light and electron microscopy (cryo-CLEM).
In cryo-electron tomography, frozen-hydrated samples are exposed to the electron beam to obtain images of the samples. The sample is physically tilted through incremental stage movements and this rotation allows for collection of different views used later to construct 3D maps, the tomograms. The concept is different to single particle cryo-EM methods [discussed in CryoEM 101], in which the grid retains the same orientation during data collection, and 3D information is obtained by combining tens of thousands of 2D images of the particles in different random orientations computationally to obtain a 3D volume.
The electron beam penetrates the sample and electrons are collected and registered at the detector. The sample is tilted by a few degrees and a new image is collected. The images collected are typically collected in a range from negative to positive 50-70 degrees (where 0 degrees is untilted) using tilt schemes with the aim to minimize radiation damage and maximize high resolution data. The combined set of images collected is referred to as ‘tilt series’ and provides many different views from the same sample. For example, a tilt series collected from -60 to +60 degrees in 3 degree increments, produces 42 images of the sample. Each image depicts a differently tilted view of the same object and images are related to each other by a known rotation allowing 3D information to be derived. The tilt-series is aligned and reconstructed as a 3D volume referred to as a ‘tomogram’.
Although 3D information can be obtained, there are challenges in cryo-ET that limit achievable resolution. For example, the same area is imaged repeatedly and therefore the total electron dose the sample is exposed to must be balanced to minimize radiation damage while generating sufficient contrast within each image. In addition, due to stage movements in between exposures and other factors, the region of interest recorded between images can be shifted, which needs to be corrected before a 3D reconstruction can be obtained. This is achieved by iteratively aligning images to each other either using fiducial markers, landmark features of the samples itself or correlation.
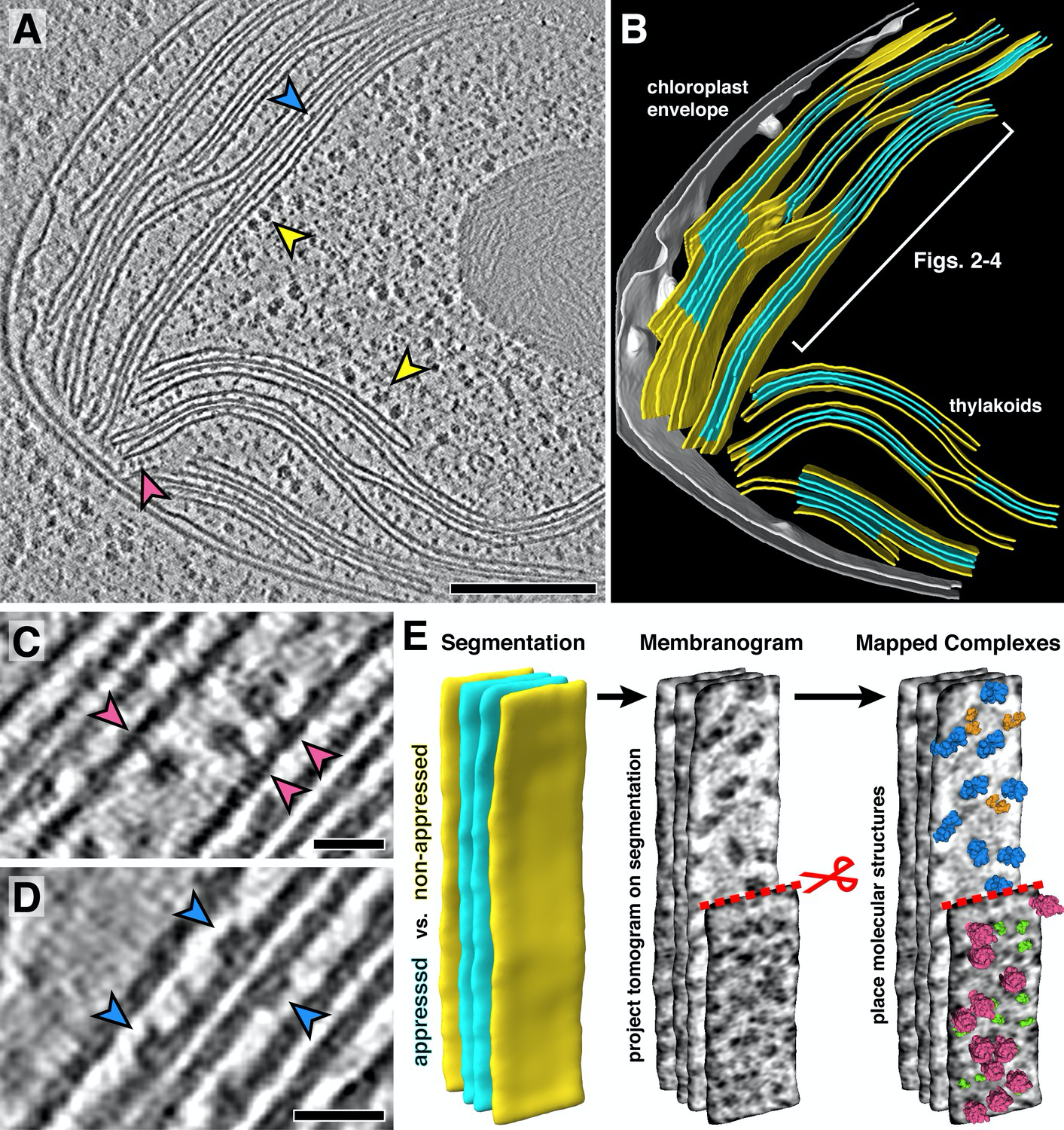
A notable limitation is the ‘missing wedge’ problem, which makes the structural information obtained from a single tomogram insufficient to obtain a structure. During data collection, not all angles of the sample are collected, since the sample becomes too thick to be penetrated by the electron beam at high tilt angles leaving an area of 3D space unimaged. These views are missing in the 3D reconstruction and appear blurry and stretched out, since there is no information available. Sub-tomogram averaging can overcome this problem (see below) and other strategies will be discussed in detail chapter 6.

Once a 3D reconstruction is obtained, the biological question determines the further workflow. If for example, large protein complexes and their location within the tissue or organelle or distinct features in a cell, like membrane locations are desired, annotation is a very helpful tool. The ever growing field of AI has yielded applications that have become tremendously helpful in tracing important scaffolds and structures in tomograms to identify and annotate biological structures like membranes, vesicles, ribosomes and proteins.
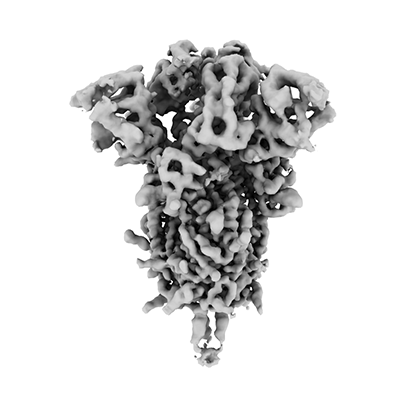
But data in tomograms can be evaluated beyond the ultrastructure: Molecular structures can be determined from proteins and their complexes in their native environment using a method called subtomogram averaging. Subvolumes containing the individual targets are selected and excised and through an iterative process of subtomogram averaging aligned to provide high resolution 3D maps. Each subvolume has a different ‘missing wedge’ owing to differences in the orientations of subvolumes within the specimen, so the lacking 3D information can be filled in. Typically, resolution of structures obtained by subtomogram averaging range around 15 Å, but with advanced processing methods and high subvolume quantity near atomic resolutions can be obtained.
Cryo-ET can be used to obtain ultra structural organization of whole organisms, viruses and their surfaces, structures of ribosomes, large complexes, organelles and their membrane proteins to name a few. Remarkably, structures obtained through subtomogram averaging from in situ tomograms can achieve near atomic resolution.
To give an overview of how cryo-ET can determine large macromolecular structures in situ, we will look at three selected examples using cryo-ET applications and the benefits provided by the technique.
For some molecular machines, there are stark structural differences observed between purified samples used in traditional structural biology methods in comparison to the same machines in their native, cellular environment. For example, a comprehensive study by Zimmerli and colleagues obtained structures of nuclear pore complexes (NPCs) directly from vitrified Saccharomyces cerevisiae cells byusing cryo-ET of FIB-lamellae. NPCs were selected from tomograms andstructures obtained through subtomogram averaging, which revealed that the pore size can be altered by tension and hypertonic shock (DOI: 10.1126/science.abd9776). These results highlighted that previously published models of NPCs were in the most constricted state, most likely caused by purification of these complexes prior to their imaging. Removal of the complexes from their native membrane environment likely promoted their constriction. Studies looking at super complexes in situ have fundamental impact. Notably, for this study, the observation of larger pores suggests that HIV can indeed enter the nucleus – since it now ‘fits’: the native pore is sufficiently wide.

This single exemplary study demonstrates the power of cryo-ET: complexes can be assessed in their native environment that may provide constraints essential to function, allowing for snapshots of functional systems.
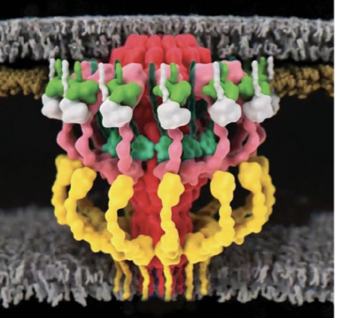
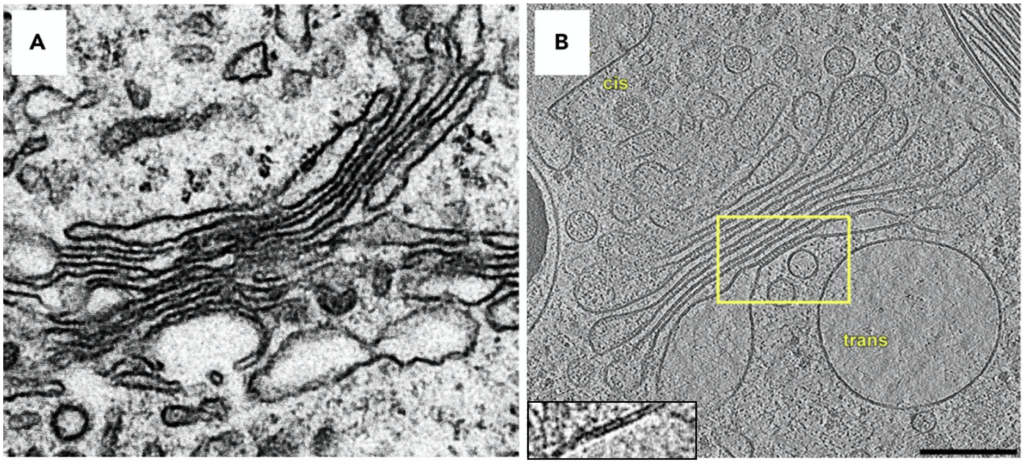
The surface (S)-layer coats the outside of bacteria in a paracrystalline-like array and is a common feature for all bacterial and archaeal cells. Proteins comprising this layer, e. g. RsaA, are known to anchor to lipopolysaccharides on the bacterial cell surface, but it was not known how they are organized or link to cells on a molecular level. Recently, Kügelgen and colleagues used a combined structural approach including cryo-ET to determine the full length structure of RsaA, define the architecture of the S-layer and how the layer is anchored to lipopolysaccharides on the outer membrane in bacteria (https://doi.org/10.1016/j.cell.2019.12.006).
This study is a powerful example of the use of cryo-ET to tie together individual high resolution structures of components obtained from cryo-EM and x-ray crystallography with native large scale assemblies by directly visualizing the architecture of a native S-layer in cellular context.
The group obtained a crystal structure (2.7 Å resolution) of the C-terminal (outermost) part of the S-layer protein RsaA from Caulobactor crescentus that spanned the C-terminal region. The full length structure forming hexamers was determined by cryo-EM (3.7 Å resolution). Tomograms of C. crescentus revealed the overall arrangement of RsaA in the surface layer and, in addition, subtomogram averaging resolved hexameric subunits (4.8 Å resolution) and how they bind to lipopolysaccharides. The study highlights the power of cryo-ET in understanding fundamental biological concepts by directly visualizing complexes and molecular assemblies in situ.

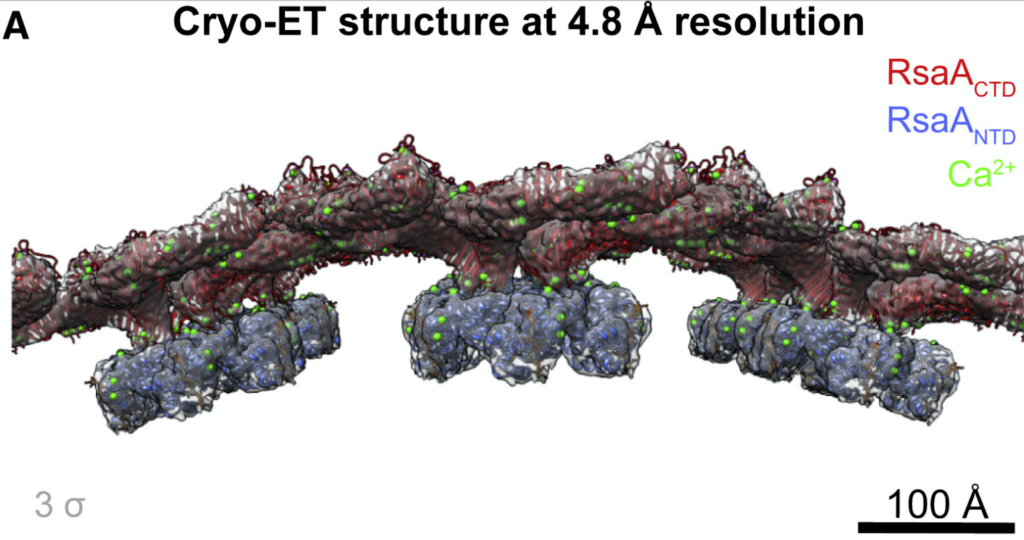
Notably, constant advances in subtomogram averaging processing approaches keep providing more detail and pushing the resolution limits. For the s-layer proteins, using the new toolkit in Relion 4 (https://doi.org/10.1101/2022.02.28.482229) focused on a bayesian approach, the map resolution could be improved to 3.5 Å. The new map revealed Ca2+ binding sites and an additional lipopolysaccharide binding site. The improvements on the data collection side and rapid development in data processing bring high resolution structures obtained through subtomogram averaging from in situ tomograms within reach.
The molecular mechanism responsible for rhythmic waves during the beating of eukaryotic flagella was determined by authors Lin and Dicastro using cryo-ET (DOI: 10.1126/science.aar1968). In eukaryotes, beating of cilia and flagella is crucial for driving motion of cells such as spermatozoa or for movement of fluids. These whip-like organelles are composed primarily of microtubules and multiple dynein isoforms that drive their rhythmic movement.
In the study, sea urchin sperm cells were vitrified and directly imaged using cryo-ET. Extensive subtomogram averaging and classification of dyneins and their regulators showed these complexes to adopt different conformations dependent on their activity during flagellar beating. These observations allowed the authors to define an intricate “switch-inhibition” mechanism that modulates flagellar motility. This study serves as an excellent example on how tomography experiments in situ can be used to reveal fundamental biological mechanisms.
The class averages obtained from different flagella regions are ranging at resolution limits of 30 Å, which is a more modest resolution. However, it facilitates confident placing of individual components and domains which helps define the molecular mechanism. The dynein active and inactive states are recognizably distinct from each other. Since the data was taken from in situ samples, the subtomogram averages of the different states could be related back to their original position along the flagella, which gave rise to a different mechanism than that was prevalent in the field.
This is a wonderful example on how in situ cryo-ET of biological specimens, entire functional sperm cells frozen during movement, can help understand fundamental biological mechanisms on molecular level driving the rethinking of established hypotheses.
The table below provides examples of several projects that have employed cryo-ET alone or in combination with cryo-EM and subtomogram averaging (STA). The studies highlighted have been selected to demonstrate the broad applicability of the method and the specific scientific questions to which it has been applied. The examples are intended to serve as a starting point to think about the possibilities cryo-ET as a method provides.
© Copyright 2024