

Cryo-EM has a distinct advantage over other structural biology methods such as X-ray crystallography and NMR in that only a relatively small amount of sample is required. Although cryo-EM can tolerate low purification yields and certain degrees of impurity, the highest quality 3D reconstructions are generally derived from samples that have gone through extensive biochemical optimization.
In this chapter, we will cover basic principles during sample purification that can make or break a cryo-EM project. Special considerations need to be taken into account based on the nature of the particle, such as its size and structural heterogeneity. The chapter introduces common considerations and strategies that are used to optimize particles for structure determination. The chapter then concludes with a negative stain section that includes protocols of how to prepare negative stain grids and how to evaluate their images. By the end of this chapter, users should understand general guidelines to assess particle quality via negative stain TEM in preparation for cryo-EM.
A successful cryo-EM project is dependent on the ability to reconstruct 2D projections of a particle into its 3D structure. The particles should meet certain criteria even before delving into cryo-EM. Most importantly, the particles should be monodisperse in solution, meaning that the sample solution consists of molecules that are roughly the same mass, shape, and size. The following section describes a few strategies that are used to improve particle behavior for single particle analysis.

1. Size exclusion chromatography: Gel filtration can be a powerful tool to predict structural heterogeneity. Whenever possible, gel filtration should be incorporated to the final steps of a purification protocol. Elution profiles can reveal the degree of monodispersity, including whether particles have tendencies to aggregate (i.e., large peaks in void volume) or whether multiple protein peaks are observed during elution. The shape of the peak is also an important indicator of monodispersity and ideally the sample produces a symmetric profile free of shoulders or tails.


2. Protein or protein complex stabilization: If negative stain or cryo-EM analysis reveals significant structural heterogeneity, then the user may consider additional methods to stabilize a structural target in a defined conformation. These approaches have proven to be effective in trapping conformationally variable proteins and complexes in energetic minima that allow high-resolution structure determination. Some examples are shown in the table below and include using small molecule inhibitors, non-hydrolyzable nucleotides (if applicable), Fab antibody fragments, or genetic perturbations (e.g., mutating catalytic residues).
| Particle | Resolution (Å) | Stabilization method | EMDB Accession No. |
| β-galactosidase | 1.9 | Small molecule inhibitor (PETG) | EMD-7770 |
| Vps4 | 3.2 | Non-hydrolyzable nucleotide analog (ADP·BeFx) | EMD-8887 |
| Ribosome Quality Control Complex | 8.2 | Catalytic inactive mutant | EMD-6170 |
| Insulin degrading enzyme | 3.7 | Fab antibody fragment | EMD-7062 |
| ABCG2 | 3.1 | Catalytic inactive mutant | EMD-0190 |
| IGPD2 | 3.1 | Small molecule inhibitor (C384) | EMD-3999 |
| CLC-K channel | 4.0 | Fab antibody fragment | EMD-8454 |
| MERS ectodomain trimer | 3.6 | Fab antibody fragment | EMD-8784 |
| HIV-1 envelope trimer | 3.8 | Fab antibody fragment | EMD-9294 |
| YME1 | 3.4 | Catalytic inactive mutant | EMD-7023 |
| Isocitrate dehydrogenase | 3.8 | Small molecule inhibitor (ML309) | EMD-8193 |
| Lactate dehydrogenase | 2.8 | Small molecule inhibitor (GSK 2837808A) | EMD-8191 |
| Dicer-2 bound to dsRNA | 6.8 | Non-hydrolyzable nucleotide analog (ATPγS) | EMD-7290 |
3. Chemical crosslinking: One common way to counteract structural heterogeneity is to chemically crosslink the purified sample, such as with glutaraldehyde or BS3. However, crosslinked samples run a risk of producing structural artifacts and this approach should therefore be considered only after a control, non-crosslinked sample has been evaluated.
To crosslink samples in a controlled manner, Holger Stark’s group developed the GraFix (Gradient Fixation) approach that reduces problems associated with particle heterogeneity. GraFix is performed using ultracentrifugation of samples through a density gradient that contains increasing concentrations of crosslinking reagent. Heterogeneous particles will separate through the gradient based on their masses and become stabilized through crosslinking. Crosslinkers should be used at low concentrations to minimize non-specific intermolecular crosslinks (e.g., 0.01-0.1% v/v glutaraldehyde may be a good starting point). Following ultracentrifugation, the tube is fractionated and samples can be assessed by SDS-PAGE and negative stain TEM. Before preparing cryo-EM grids, the gradient material (e.g., glycerol or sucrose) may interfere with vitrification or decrease contrast in cryo-EM images and should thus be removed via buffer exchange.
Membrane proteins are an increasingly popular class of macromolecules for single particle cryo-EM analysis. These types of proteins are important targets of pharmacological agents but are challenging to crystallize and study by other structural biology methods. There are now many examples demonstrating that cryo-EM is ideally suited for resolving high-resolution structures of membrane proteins. In order to visualize membrane proteins, they are either solubilized in the presence of detergents or detergent-like amphipols, or reconstituted in lipid nanodiscs.
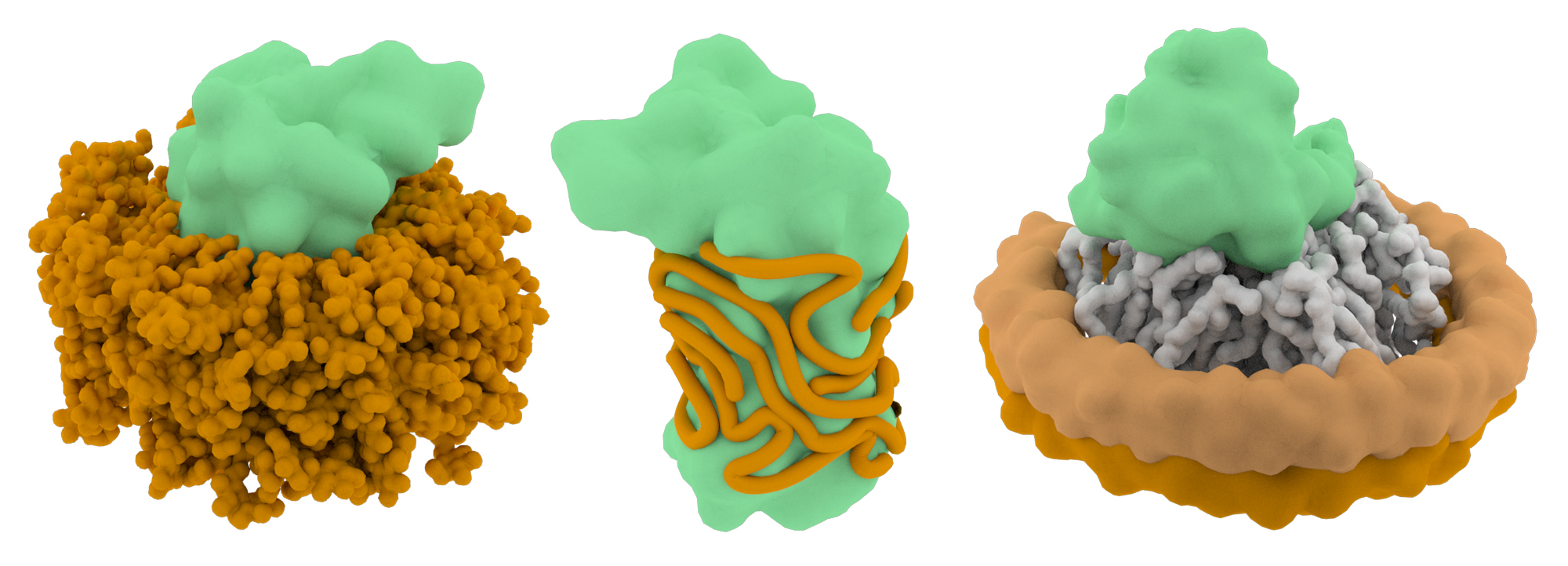
Detergents are required to solubilize membrane proteins, but the molecules may also destabilize their structures. The choice of detergent is therefore a critical part of the project workflow and multiple types may need to be evaluated before one that strikes the right balance between solubility and structure preservation is determined. The concentration of the detergent is also a variable that affects particle quality. Most detergent-stabilized membrane protein structures are solved using detergent concentrations at or below the critical micelle concentration (CMC), and this range may be a good starting point for single particle EM projects. However, keep in mind that detergents may complicate the cryo-EM grid preparation process (by affecting the surface tension of the remaining solution on the blotted grid) and detergent molecules may lower the contrast of cryo-EM images.
An attractive alternative to detergents is to use amphipols to stabilize membrane protein structures. Amphipols are long, amphipathic polymers (~8-9 kDa) that can be used to replace detergents in buffer. The hydrophobic chains wrap irreversibly around transmembrane regions, forming a tight belt that shields the hydrophobic domains from the aqueous environment. The detergents are then removed from solution via Bio-beads or dialysis. Most of the current high-resolution membrane protein cryo-EM structures have been solved using amphipols.
One emerging tool to study membrane proteins via single particle EM is the use of lipid nanodiscs that stabilize the proteins in a lipid bilayer environment. These nanodiscs consist of a patch of lipid bilayer that is surrounded by a belt of membrane scaffolding proteins. The major advantage to using lipid nanodiscs is that the proteins are retained in more a native-like environment. Thus, lipids that directly interact with membrane proteins are also retained and resolvable by cryo-EM. Although nanodiscs are the most desirable choice to study membrane protein structure, there is currently no ‘one-size-fits-all’ system that allows the nanodiscs to form. The size of the membrane scaffolding proteins often needs to be optimized to form the lipid belt properly and their successful assembly can be validated by size exclusion chromatography.
The following table provides examples of several membrane protein structures that have been solved using one of these three different methods (detergents, amphipols, or nanodiscs). Keep in mind that the right choice for your protein-of-interest will probably require extensive trial-and-error and may be the rate-limiting step of your project.
| Membrane Protein | Resolution (Å) | Stabilization method | EMDB Accession No. |
| TRPV1 | 3.3 | Amphipols | EMD-5778 |
| TRPV1 | 3.3 | Nanodisc | EMD-8118 |
| PKD2 | 3.0 | Nanodisc | EMD-8354 |
| ABCG2 | 3.6 | Nanodisc | EMD-0196 |
| Kv1.2-2.1 | 3.3 | Nanodisc | EMD-9024 |
| TMEM16A | 3.8 | Nanodisc | EMD-7095 |
| TRPM4 | 3.1 | Nanodisc | EMD-7133 |
| TRPV6 | 4.0 | Amphipols | EMD-7121 |
| STRA6 | 3.9 | Amphipols | EMD-8315 |
| TFP | 4.2 | Detergent (DDM) | EMD-6945 |
| Orco | 3.5 | Detergent (digitonin) | EMD-7352 |
| γ-secretase | 3.4 | Amphipols | EMD-3061 |
| Class B GPCR-G-protein complex | 4.1 | Detergent (LMNG/CHS) | EMD-8623 |
| RyR1 | 3.6 | Detergent (CHAPS) | EMD-8342 |
| CFTR | 3.9 | Detergent (digitonin) | EMD-8516 |
Particles must be visible under the electron microscope in order to be properly analyzed. This constraint poses a lower molecular weight limit of what can be a viable cryo-EM project. If the particle is too small, it may be entirely invisible in the cryo-EM micrograph. Even if the particle is visible, its small size may lead to inaccurate orientation determination during downstream image processing steps and produce unreliable structures. For practical purposes, a general guideline is that particles should be at least 100 kDa in size, although the true lower limit is unknown.
How do you know that your purified sample is good enough for single particle cryo-EM? Virtually every cryo-EM project begins with negative stain analysis to evaluate particle quality. This simple and quick method provides a first-order estimation of the particle’s monodispersity, concentration, and averageability.
Protocol
Table of common reagents used for negative stain TEM
| Stain | Working concentration | Comments |
| Uranyl acetate | 1-2% | Most common stain used for single particle analysis. Light sensitive. Store at 4°C. Filter periodically to remove precipitates. |
| Uranyl formate | 0.5-1% | Light sensitive. Difficult to dissolve. Store single use aliquots at -80°C. The fine granular size of this stain is useful for visualizing smaller particles (<100 kDa). |
| Phosphotungstic Acid | 1-3% | Prepared at neutral pH and may be preferred if particles are sensitive to acidic stains. Provides less contrast than uranyl-based stains. |
| Ammonium molybdate | 1-5% | Prepared at neutral pH and may be preferred if particles are sensitive to acidic stains. Provides less contrast than uranyl-based stains. |
Negative stain images should be recorded at a few microns underfocus. Optimally stained areas of the grid should show fine details within individual particles. The following interactive module shows a few negatively stained bacteriophage particles. Use the module to adjust the focus parameter of the images. Note how the images become blurry when you’re far from focus. For negative stain particles, 1 to 2 microns of underfocus generally provides a good balance of detail and contrast in the final images.

Ideally, particles should be well dispersed, not aggregated. There should be enough particles to saturate the field of view because cryo-EM samples generally show at least 5-10x fewer particles than negative stain. Particles that are well stained will show textured features. Samples that are poorly stained or overstained will appear as dark blobs with no identifiable features.

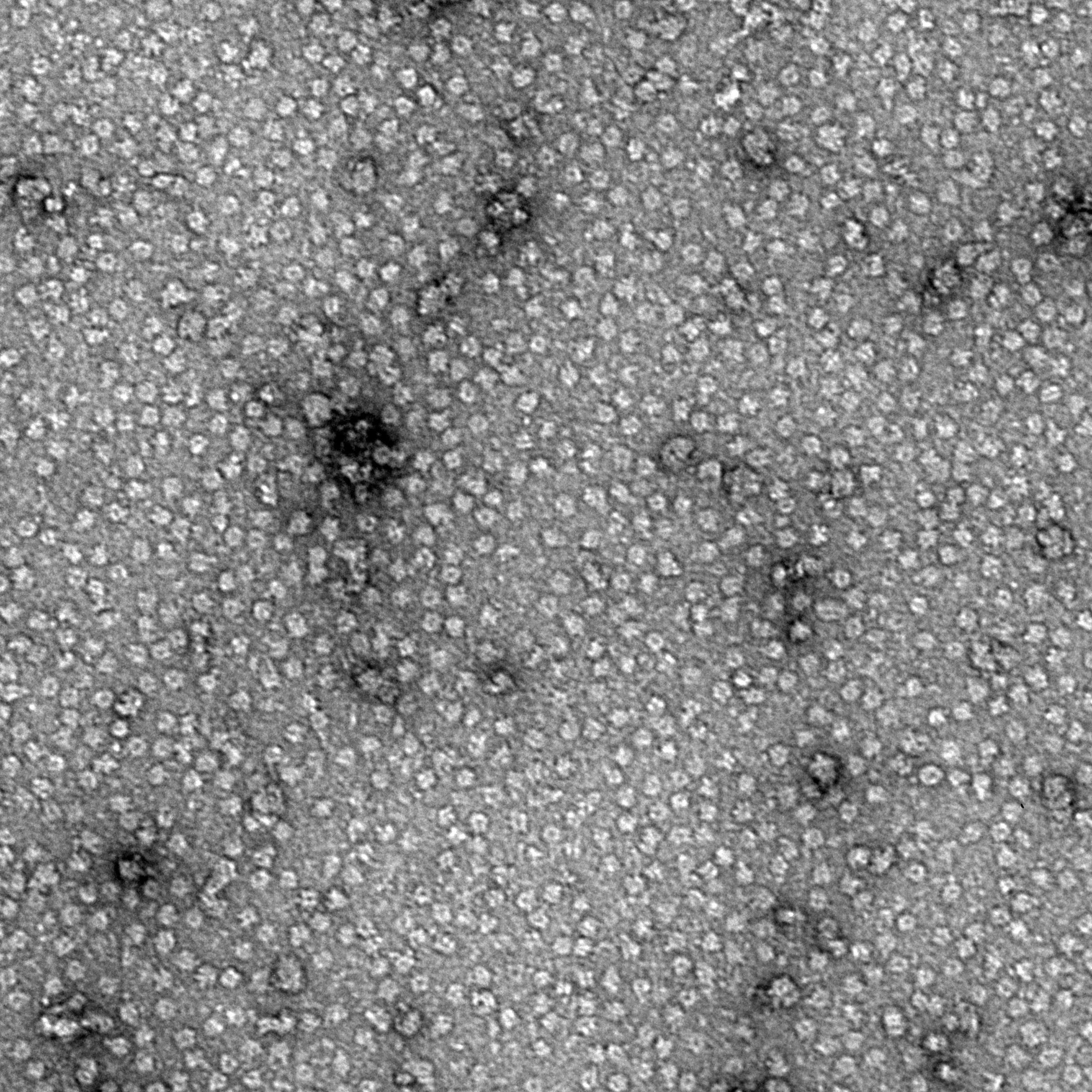
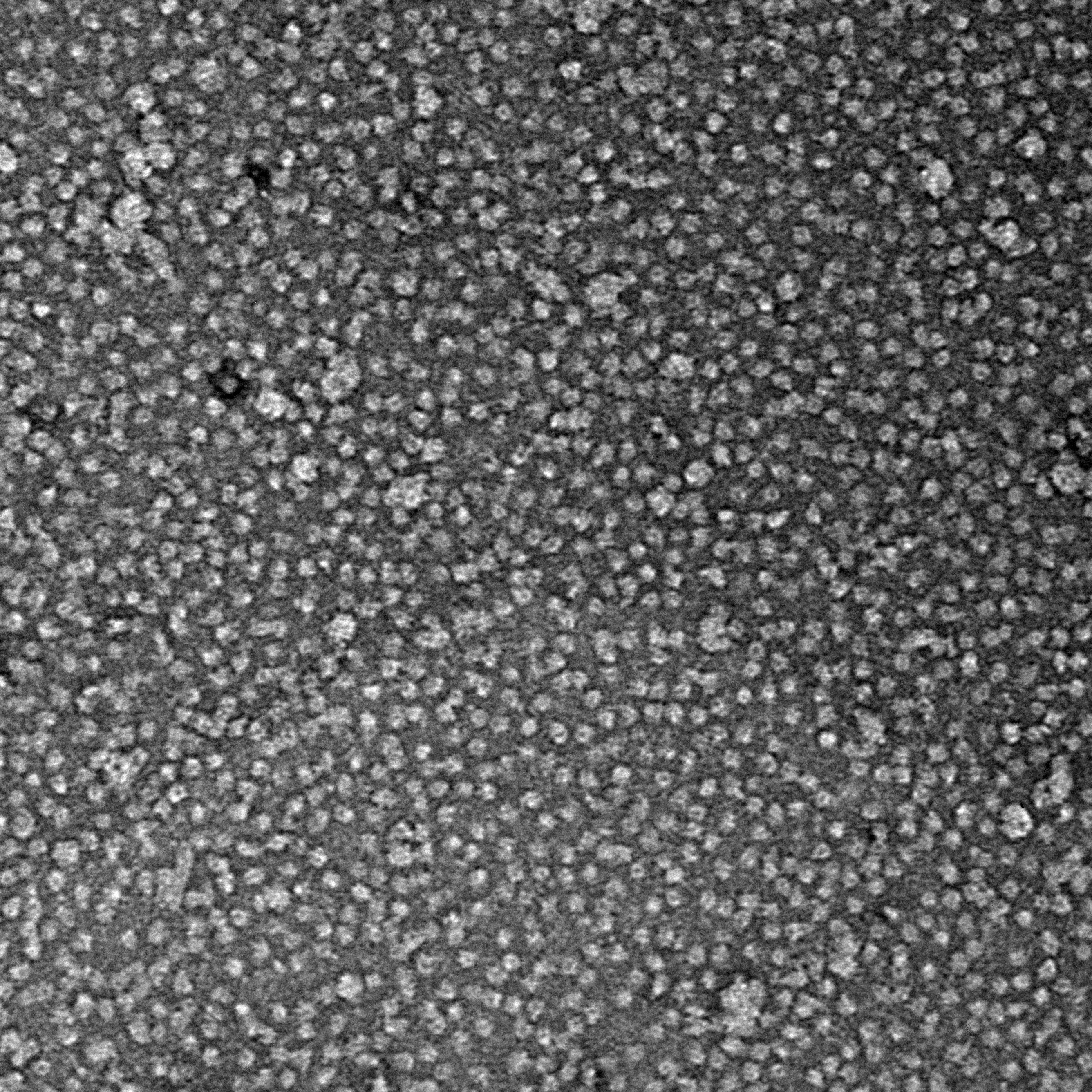




Negative stain images can be used to provide a sense of how well the particles could be averaged. In this procedure, individual particle images are extracted from the micrographs and then sorted into separate classes based on their similar features. A dataset of a few thousand particles is usually sufficient for this exercise. Attaining well-averaged 2D classes is a good indicator that the particle is ready for cryo-EM and is a minimal requirement expected by many cryo-EM facilities. A more detailed overview of 2D class averaging is available in Chapter 5 (Data Processing).
Note that negative stain analysis is not always an accurate predictor of whether the particle will lead to a high-resolution cryo-EM structure. Proteins could be destabilized by different stains, for example due to the acidic pH of some stain solutions. Small particles may be harder to visualize by negative stain than by cryo-EM because of poor interactions between the stain and particle. Nucleic acids can also be challenging to visualize by certain types of negative stain. Other issues with single particle cryo-EM, such as preferred orientations and particles failing to enter holes, cannot be addressed at the negative stain level. Nevertheless, it is always worth performing negative stain analysis because of the quick feedback it provides.
Mass photometry is an emerging method that provides information about particle mass and monodispersity of biomolecules in solution. A small volume of sample is deposited onto a glass surface, which is then illuminated by a laser. The light is reflected off the glass surface and is also scattered by the particles on the surface. The scattered and reflected light interfere with each other, which is captured by an objective. The amount of interference from each biomolecule scales linearly with its mass, independent of shape or volume. Mass photometry can measure the masses of biomolecules (protein, nucleic acid or other biomolecule of interest) ranging between 30 kDa to 5 MDa in mass.
A number of features of mass photometry make it suitable for a cryo-EM workflow. Mass photometry is compatible with most aqueous buffers and detergents, so it is possible to analyze biomolecules, including membrane proteins and macromolecular complexes, in their native states. The method offers a fast readout time and has low sample requirements, thus making it a desirable technique to screen different buffer conditions before proceeding to negative-stain EM and cryo-EM.
Mass photometry is a single-particle technique, so it can also detect rare species in a sample. These attributes allow mass photometry to provide better resolution over heterogeneous samples than techniques which rely on averaging particle masses, like dynamic light scattering (DLS). The sample concentration needed for mass photometry experiments is very low (10uL of sample at nanomolar concentration), which conserves valuable sample during early optimization steps; however, this can also be a limitation if the complex requires higher concentrations for stability or grid preparation. Sometimes even a higher concentration of the biomolecule is not enough to withstand the effects of the air-water interface or freezing. In these cases, additional stabilization from crosslinkers or fixatives may be necessary, and mass photometry can be used to optimize structure stability under these conditions.
Overall, mass photometry offers a rapid sample optimization and analysis method for cryo-EM projects and is complementary to other quality control methods, such as negative stain screening. This technique can provide information that is not otherwise accessible with other instruments and can streamline the sample characterization process before committing additional resources to cryo-EM.
© Copyright 2024
